
近日,黑龙江大学化学学院付宏刚教授、刘健聪教授和王东旭高级工程师团队在光驱动低温催化CO2加氢制CO领域取得重要进展。太阳能驱动的低温逆水煤气变换反应是实现CO2高值化利用的理想路径之一,但目前仍面临反应效率较低和反应机制尚不明确的问题。研究团队设计合成了Pt团簇负载CeO2催化剂,实现了光驱动下的表面电荷重排,解锁了双路径协同催化,显著提升CO生成速率。仅以光为能量输入(2.27 W/cm2),催化剂表面局域温度可达~309°C,而反应器内仅约54°C。在连续流动体系中,该催化剂实现了846.9 mmol·gcat-1·h-1的CO产率,性能优于同温度下传统热催化体系。在户外-21 °C环境温度下,仅利用自然太阳光仍可获得相当的催化性能。机理研究表明,光驱动表面电荷重排构建了非平衡态Ptδ+–OV–Ce3+结构,该结构促进CO2活化,激活羧酸盐路径并增强甲酸盐路径,协同加速整体反应速率,这与热催化条件下单一甲酸盐路径形成鲜明对比。本工作表明光可作为一种动态调控催化位点的有效手段,为高效太阳能转化提供了新思路。研究成果以“Light-Driven Charge Redistribution of Pt Cluster/CeO2 Realizing Dual-Path Synergistic Catalysis for Low-Temperature Reverse Water-Gas Shift”为题,发表于《Angewandte Chemie International Edition》。
采用胺辅助锚定策略,制备了高分散Pt团簇/CeO2(PtC/CeO2)催化剂。该策略有效抑制了热处理过程中Pt物种的迁移团聚,构筑了丰富的Pt–O–Ce界面结构。通过调控合成条件,还可获得单原子催化剂(PtSA/CeO2)作为参照。传统浸渍法制备的Pt粒子催化剂(PtN/CeO2)呈现不均匀的Pt粒子分布。HAADF-STEM图像表明,Pt团簇高度分散在CeO2表面,同时存在原子级分散的Pt物种。CO-DRIFTS结果显示,PtC/CeO2同时存在归属于Pt0–CO和Ptδ+–CO的两个CO吸收主峰,并伴有较弱的桥式CO信号,证实了Pt团簇与单原子Pt的共存。相比之下,PtN/CeO2呈现明显的桥式CO峰,对应尺寸较大的Pt纳米颗粒。(图1)
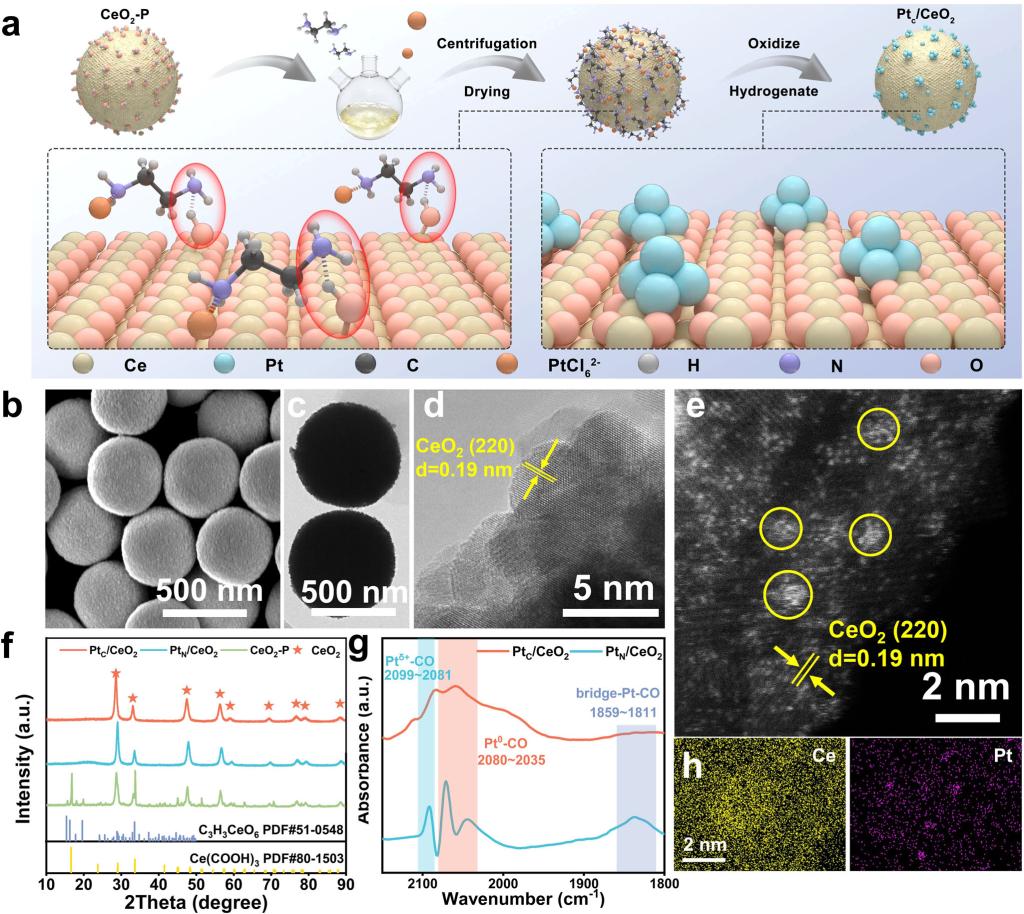
图1. PtC/CeO2催化剂的合成示意图及结构表征
XANES和EXAFS进一步揭示了Pt的局域配位环境。PtC/CeO2中Pt L3边白线峰强度位于Pt foil和PtO2参照之间,表明Pt价态在0到+4之间。EXAFS协同小波变换分析共同证实了PtC/CeO2中Pt–O和Pt–Pt配位的共存。结合Pt 4f XPS谱图进一步证实了PtC/CeO2中Pt簇和单原子Pt的共存。H2-TPR和DFT结果表明PtC/CeO2具有更大的氧空位(OV)形成潜力。XPS和EPR进一步证实,相对于Pt粒子催化剂,Pt团簇催化剂具有更高的表面Ce3+比例和OV浓度。CO2-TPD和UV-vis DRS表明,增强的OV特性显著促进了PtC/CeO2催化剂对CO2吸附/活化能力和光吸收特性,为其优异的光驱动催化活性奠定了良好基础。(图2)
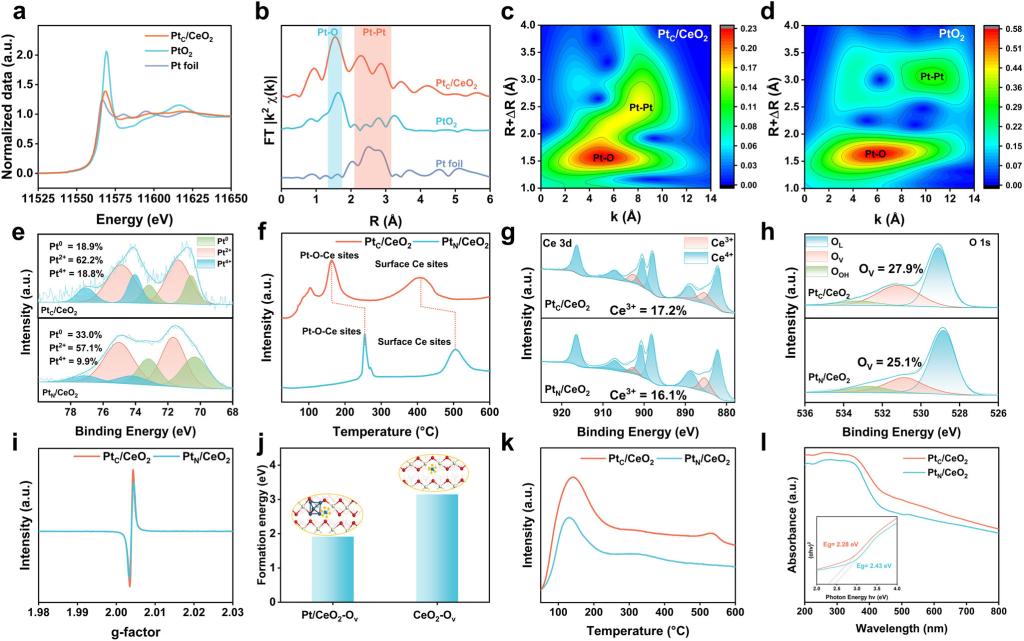
图2. PtC/CeO2催化剂的结构和物理化学性质表征。
在连续流动反应器中,以全光谱氙灯为唯一能量输入,不施加任何外部加热,测试了催化剂的光驱动RWGS性能。随着光强的增加(1.18-2.75 W/cm2),催化剂表面温度从201升高至347°C,CO2转化率从2.98%升高至33.21%,CO选择性始终保持在97%以上。在相同的催化剂表面温度下(309°C),光驱动CO2转化率可达22.20%,是传统热催化(10.82%)的2.1倍。光强为2.75 W/cm2时,CO2转化率可达33.21%,超过该温度下的热力学平衡转化率,凸显了光驱动过程的非平衡态本质。在高空速下,CO产率高达846.9 mmol gcat-1 h-1。连续122小时稳定性测试表明催化剂具有良好的催化稳定性。户外实验验证了太阳光驱动RWGS反应的可行性。尽管环境温度低至-21°C,利用菲涅尔透镜汇聚阳光驱动下,CO2转化率可达30.81%,CO选择性保持在98.26%,与实验室氙灯条件下的性能相当(33.21%),充分展示了该体系的实际应用潜力。(图3)
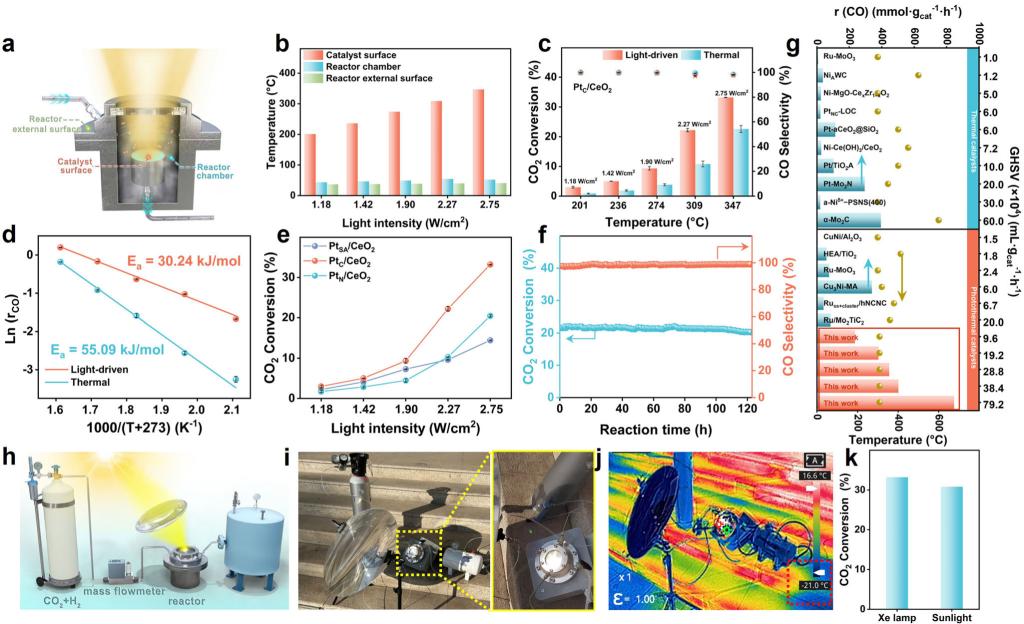
图3. PtC/CeO2催化剂光驱动RWGS性能研究以及户外模拟实验。
原位红外(DRIFTS)研究发现,在热催化条件下仅检测到甲酸盐(HCOO)和双齿碳酸盐(b-CO32-)中间体,未检测到羧酸盐(COOH)信号。而在光驱动条件下,HCOO和COOH中间体同时出现。这一发现证明光驱动过程中的双路径协同机制(图4)。进一步地,不同波段光过滤等温实验验证了光化学过程的存在:在相同温度(201 °C)下,紫外和可见光照射下的CO产率均高于纯热催化过程,证明光不仅仅提供热源,其光化学效应确实参与了反应。
利用一系列原位光谱实验揭示了光致双路径协同机制的内在因素。原位光照EPR显示,光照下氧空位特征信号(g = 2.003)增强。原位光照XANES表明,光照后Pt L3边白线峰增强(Pt失电子)、Ce L3边白线峰减弱(Ce得电子)。原位光照XPS进一步显示,光照下Ce 3d和O 1s结合能负移、Pt 4f正移,Ce3+比例从17.2%升至18.9%,氧空位浓度从27.9%升至34.9%。上述结果证实光驱动电子从Pt向CeO2转移,形成Ce3+/OV和Ptδ+结构。这一光致电荷重排原位构建了非平衡态Ptδ+–OV–Ce3+活性结构:富电子的Ce3+/OV对促进C=O键活化,协同促进了羧酸盐和甲酸盐路径并行高效推进,实现反应活性的协同提升。(图4)
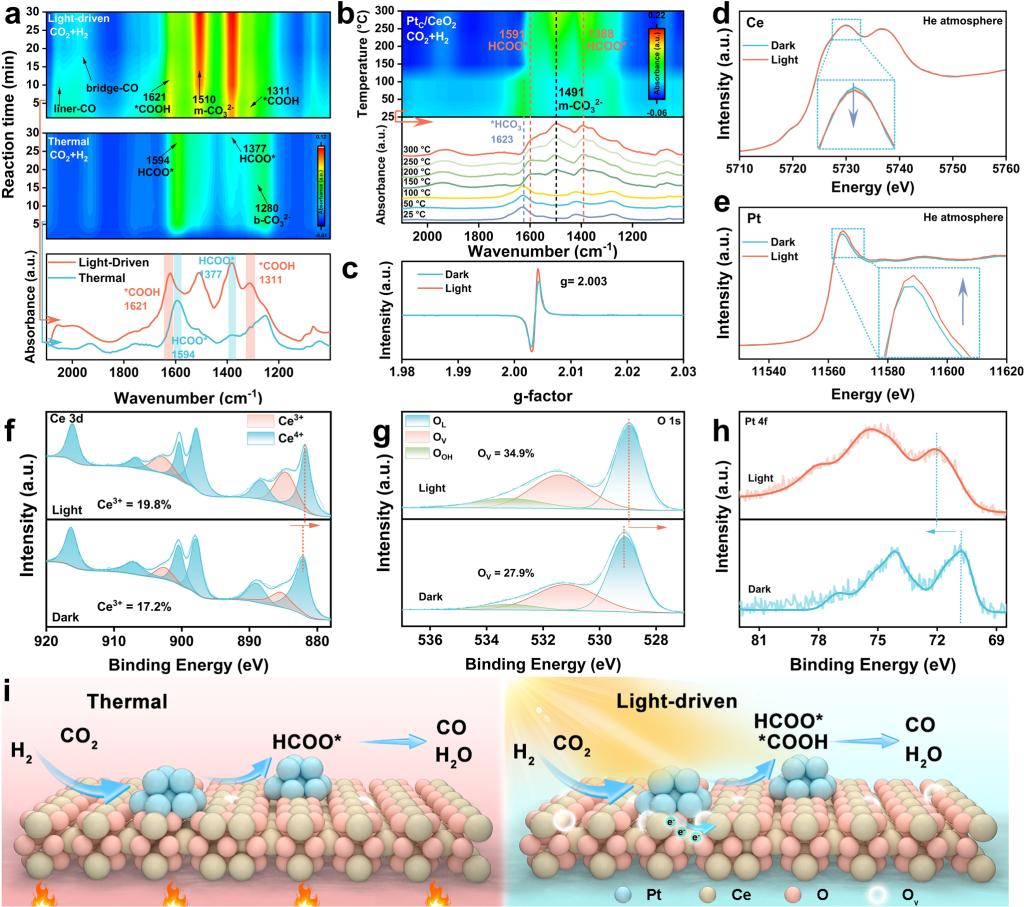
图4. PtC/CeO2催化剂的原位光谱表征以及光驱动反应机制。
综上所述,本工作通过胺辅助锚定策略构筑了高分散Pt团簇/CeO2催化剂,揭示了光驱动界面电荷重排构建非平衡态Ptδ+–OV–Ce3+活性中心,实现甲酸盐与羧酸盐双路径协同催化的新机制。该体系仅以光为能量输入,在连续流动反应器中实现了低温高效RWGS反应,CO产率达846.9 mmol gcat-1 h-1,选择性近100%,并在户外-21 °C自然光条件下获得与实验室相当的催化性能。该工作以RWGS为模型反应,建立了光驱动动态界面调控催化反应路径的新范式,为太阳能驱动的绿色低碳转化提供了可借鉴的设计策略。
黑龙江大学硕士生黄贵宇、中国科学院上海应用物理研究所乔盼哲为论文共同第一作者。通讯作者为黑龙江大学付宏刚教授,刘健聪教授和王东旭高级工程师。研究工作得到了国家重点研发计划、国家自然科学基金等项目的支持。
论文链接:
https://doi.org/10.1002/anie.5591194(化学化工与材料学院供稿)



